Cancer Research: Dendritic Cell Vaccines
STEP ONE
Be informed. Read information below.
STEP TWO
Schedule consultation with R. Douglas Wichman, MD.
STEP THREE
Submit "New Patient Form", current labwork, & imaging results prior to consultation.
STEP FOUR
Establish if you are a candidate for this therapy
in accordance with the Georgia
"Access to Medical Treatment Act".
----------------------------------------------------------------------------------------------------------------------------------------------------------
*** SPECIAL NOTE FROM DR. WICHMAN ***
The following excellent article was reproduced from Neurosurgery Clinics of North America at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2810429/:
Dendritic Cell Vaccines for Brain Tumors
Introduction
Dendritic cells (DC) have long been regarded as the most potent antigen presenting cells (APCs) within the immune system. Their ability to sample environmental antigens and stimulate T cell activity in a major histocompatibility complex (MHC)-restricted manner has attracted much attention given the poor antigen presenting ability and immunogenicity of tumor cells [1,2]. Although DCs constitute approximately 0.3% of all circulating blood leukocytes, they serve as the sentinels of the immune system and are found nearly ubiquitously throughout the body [3]. In their immature state, DCs are highly specialized antigen samplers capable of surveying their microenvironment through several mechanisms including engulfment, macropinocytosis, and receptor mediated endocytosis [3]. Upon encountering an antigen the DC processes it through MHC pathways and directs it to the cell surface to form an MHC-peptide complex (Figure 1). In line with traditional antigen presentation following uptake from the environment, many antigens are channeled through MHC-class II pathways with resultant MHC-peptide complexes being capable of stimulating CD4+ T cells. In addition, dendritic cells possess the unique ability to “cross-present” acquired antigens. In this process, DC endosomes release captured antigenic material into the cytosol where it is broken down by proteasomes [4]. The degraded peptides are then transported to the ER via a transporter-associated protein (TAP) and bound to MHC-class I molecules for presentation to CD8+ T cells [5,6]. These distinct mechanisms allow DCs to stimulate T cells in an MHC-class I and II manner, overcoming classical restrictions in antigen processing and presentation [7] and diversifying the resultant immune response.

Dendritic cells are capable of handling a vast range of antigenic mediums. The sources of antigen that have been used in DC immunotherapy include exogenous MHC-restricted peptides, acid-eluted tumor peptides, tumor RNA and cDNA, viral vectors, apoptotic tumor cells, tumor cell lysate, and whole tumor cells. Many of these methods have been employed with varying degrees of success. However, a growing sentiment has emerged that argues for the use of a diverse range of antigens that cover both MHC classes rather than constructing specific MHC-matched peptides. The reasoning for this is multifold. First, stimulating T cells with a broad range of antigens reduces the likelihood of an escape phenomenon in which tumor cells lacking the specific antigens of interest avoid immune detection and continue to grow unhindered. Second, it is now well established that the stimulation of both CD4+ and CD8+ T cells is crucial in the activation and maintenance of anti-tumor immunity [7–10]. By allowing DCs to present and cross-present antigens on MHC-class II and I molecules, respectively, one avoids having to laboriously engineer peptides for each MHC class [9,11]. Finally, the methods employed to load the spectrum of antigens for a particular tumor obviate the need of characterizing each individual antigen used. Although the use of unfractionated tumor material containing unknown antigens has long raised the concern of inducing auto-immunity, particularly in the form of experimental allergic encephalomyelitis (EAE), no reports of this complication have been seen following DC vaccination in humans to date [3].
A dendritic cell vaccine is defined as DCs loaded with antigens, for example, those found on glioma, which are administered to patients in order to induce an antigen-specific T cell mediated anti-tumor response [12]. However, though immature dendritic cells are not functionally ideal for the loading of antigens, they are unable to activate lymphocytes until an inflammatory signal or pathogen induces their maturation [3,9,11]. Some groups argue that ex vivo maturation of DCs through CD40L or interferon (IFN)-γ [13] is thus necessary prior to vaccine administration to ensure proper antigen presentation and T cell activation [14–17]. Others maintain that maturation occurs naturally, and that no prior stimulus is required [18]. In the process of maturation, DCs lose their ability to uptake and process antigens. Moreover, they exchange their immature molecular signature for a mature (CD83+) phenotype, increasing expression of MHC-antigen complexes, lymphocyte costimulatory molecules (e.g. CD80/B7-1 & CD86/B7-2), TNF and TNF-receptor molecules (e.g. CD40), and many chemokines and chemokine receptors (e.g. IL-12, IL-15, IL-18) to aid in T-cell recruitment and DC navigation to lymphoid tissues (as reviewed by Steinman [11] and Soling [9]).
Upon localization to lymph organs rich in naïve T cells, mature DCs present their processed antigens in a MHC-restricted manner. Through various interactions they are able to mobilize many different arms of the immune system, including CD8+ cytotoxic T cells (CTLs), CD4+ helper T cells, natural killer (NK) and NK-like T cells [11]. Each of these cell types plays an essential role in the anti-tumor response (Figure 2). T cells expressing CD8 co-receptors recognize and lyse tumor cells in an MHC-class I restricted fashion, and have received much of the credit as the primary effector cell in immunotherapy. CD4+ T cells have traditionally been known for their part in the expansion and maintenance of CD8+ CTLs, secretion of stimulatory cytokines, and the induction of lasting immunity. Their critical role in immunotherapy has become increasingly appreciated over the past few years, as studies have demonstrated that their absence may result in deficient DC maturation as well as CTL tolerance [7,8]. Finally, NK and NK-like T cells have a unique niche in the leukocyte armament, being able to recognize and kill tumor cells that do not express surface markers such as MHC-class I. Although the exact mechanism of recognition and elimination of tumor cells in the absence of MHC-restriction is yet to be elucidated, they serve as an important complement in the killing of tumor cells that may possess diminished surface marker presentation and avoid CTL detection [3,11].

Animal Models
Pre-clinical animal models explored many of the methodologies and safety concerns regarding dendritic cell vaccines. In the late 1990’s, we were among the first to describe the effectiveness of dendritic cell vaccines in the anti-tumor immunity of gliomas using a rat model [19]. Over the past decade, many groups have published similar studies with varying permutations in the choice of antigen, timing of vaccinations, and measures of therapeutic efficacy. The dissimilarities between study designs make it difficult to compare methodologies and their associated outcomes. However, their value lies within their ability to demonstrate the effectiveness of multiple different DC vaccine techniques in inducing antigen-specific cytotoxicity in vitro and in vivo. These studies have shown improved survival outcomes, as well as the safety of the strategy.
Many of the initial animal studies were key in evaluating the effectiveness of DC vaccination techniques for tumors located in the “immunologically privileged” central nervous system (CNS). Antigen sources used included synthetic peptides [20,21], acid-eluted tumor peptides [19], tumor lysate [21–26], DC-tumor fusion cells [27], and antigen containing vectors such as cDNA/RNA carrying viruses [28,29] and tumor extract carrying liposomes [30]. Many of these studies adopted strategies for antigen loading of DCs from previous experiments in peripheral neoplasms, and initial treatment schedules were similarly based on regimens that showed promise in non-CNS immunotherapy[19]. Nevertheless, the timing of DC administration variedly widely, with vaccinations being given before tumor inoculation [22,24,26–28], simultaneously with [21,23,25], or some time following tumor implantation [19,24,27,28,30]. The number of vaccinations ranged from two to five times across the various studies.
Overall, most groups concluded that vaccination with antigen-pulsed dendritic cells was able to produce a significant anti-tumor immune response. This was evidenced by increased overall survival in rats and mice, greater degrees of T-cell infiltration (primarily CD8+) on histological analysis of tumors, and more robust anti-tumor cytotoxicity assays when splenocytes were incubated with mouse glioma in vitro in immune responders. Although the studies conceded that pre-tumor vaccination resulted in greater survival in animal models, there are conflicting reports regarding the efficacy of DC vaccines when administered simultaneously or following tumor implantation [21–25]. Groups reporting no improvement in survival with DC vaccination after an established tumor suggest that it may be due to the immune system failing to generate an appropriate response quickly enough to counteract a rapidly growing tumor within a confined cranium[21,24]. However, studies in which T-cell mediated tumor killing was achieved showed that the animals that did respond to DC vaccination obtained lasting anti-tumor memory, with significantly improved survival following tumor rechallenge compared to unvaccinated controls [21,22,26].
These animal studies played an important role in alleviating some of the concerns regarding DC immunotherapy for CNS tumors. Experimental allergic encephalomyelitis (EAE) in particular was perhaps one of the most feared side effects, as previous studies have shown this lethal form of autoimmunity to occur following the injection of glioblastoma tissue into animals [31]. However, such signs of autoimmunity were not reported in the more recent studies conducted to date [21,22]. In addition to the information they provided regarding the applicability of different antigen sources and treatment schedules, the pre-clinical reports corroborated the idea that DC immunotherapy could be effectively used for intracranial neoplasms, and thus set the stage for further clinical studies.
Clinical Trials
In 2000, we published a case report on the first brain tumor patient to be treated with DC-based immunotherapy [32]. A patient with histologically confirmed GBM received three biweekly intradermal injections of DCs pulsed with acid-eluted, allogeneic MHC-I matched GBM peptides. Although we were able to appreciate an immune response as evidenced by an increased infiltration of CD3+ T cells in post-vaccination tumor, there was no objective clinical response from the treatment. The patient’s poor Karnofsky performance score (KPS) in addition to the possible lack of antigen homology between the allogeneic GBM and the patient’s tumor may have contributed to the lack of clinical response or prolonged survival.
In a Phase I dose-escalation clinical trial, we treated 12 GBM patients using DCs pulsed with autologous acid-eluted MHC tumor peptides in a dose-escalation study [33]. Patients were separated into three cohorts, each receiving 1, 5, or 10×106 DCs per injection. Subjects tolerated the procedure well with no signs of autoimmunity. There were only minimal Grade I toxicities related to the study vaccine, which were distributed similarly across all three dose groups. Although this study was not powered to measure efficacy, patients undergoing DC-based immunotherapy appeared to have an increased median time to progression (15.5 months) and overall survival (23.4 months) compared to historical controls. Of note, tumor burden and disease progression at the time of vaccination was a critical determinant of systemic CTL activity, tumor infiltration by T cells, as well as overall survival. All patients that generated a systemic CTL response showed no MRI evidence of progressive disease at the time of vaccination. Conversely, no patient with actively progressive disease developed statistically significant cytotoxicity. Moreover, only patients possessing minimal tumor burden at the time of vaccination were found to have tumor infiltrating lymphocytes (TILs) upon post-vaccination tissue examination. These findings suggest that active tumor progression or bulky residual burden can debilitate the initiation and propagation of an anti-tumor response. Interestingly, expression of the inhibitory cytokine TGF-β2 was found to be inversely proportional to the number of TILs found in tumor tissue following vaccination (IL-10 was not), implicating TGF-β2 as a possible mediator of immune evasion following vaccination. This study argues for the need for maximal resection and/or minimal residual disease to improve the efficacy of DC-mediated immunotherapy for glioma. Importantly, this clinical trial established the feasibility, safety, and immunological potential of DC vaccines for brain tumor patients.
Yu et al. reported another Phase I clinical trial using DC pulsed with autologous, acid-eluted peptides for glioma patients [34]. In this study, nine patients with newly diagnosed malignant glioma received 3 biweekly subcutaenous injections of DCs loaded with acid-eluted tumor peptide. Systemic antitumor cytotoxicty was detected in 4 of the 7 patients assessed; intratumoral CD8+ CTL and CD45RO+ memory T-cell infiltration was found in 2 of the 4 patients who underwent a second resection due to tumor progression. Patients receiving DC vaccination were found to have an increased median survival (455 days) compared to that of those in the control group (257 days).
Kikuchi et al. used DC-glioma fusion cells (FCs) to vaccinate glioma patients in their Phase I clinical trial [35]. Eight patients with malignant gliomas received FCs intradermally every 3 weeks, with the total number of injections ranging from 1 to 9. An increased percentage of NK cells was found on FACS analysis in the peripheral blood of patients. In addition, an increase in IFN-γ release in PBMC-tumor co-incubation with both autologous as well as allogeneic glioma was seen following DC vaccination. Two patients experienced a minor response and no serious side effects were observed. These findings suggested that non-specific anti-tumor cytotoxicity may play a role in the DC-based immunotherapy of glioma.
Kobayashi et al. vaccinated five patients with autologous glioma RNA-pulsed DCs [36]. They were able to demonstrate the presence of a strong CD8+ CTL response against autologous glioma accompanied by a weaker NK cell-mediated cytotoxicity in their patients. This finding was significant in 3 of the 5 patients treated. Notably, in the two patients with minimal immune responses, a constitutively increased expression of the inhibitory cytokine IL-10 and decreased expression of IFN-γ by CD8+ T cells was found in vitro.
Yamanaka et al. compared different routes of DC injection in their Phase I/II clinical trial of 10 patients [37,38]. Dendritic cells were pulsed with autologous tumor lysate and administered to patients intradermally (n = 5) or both intradermally and intratumorally via an Ommaya (n = 5) reservoir every 3 weeks for a total number of injections ranging from 1 to 10. Immunologically, they observed an increased percentage of NK cells and increased T-cell mediated anti-tumor activity. In addition, there was an increased intratumoral infiltration of CD4+ and CD8+ T cells in the two patients who underwent reoperation following vaccination. Radiographically, the two minor responses seen were in patients included in the combined intradermal/intratumoral administration group, suggesting that the additional intratumorally injected DCs may stimulate a more efficient anti-tumor immune response.
Wheeler et al. published a report examining the correlation between thymic function, as manifest through CD8+ recent thymic emigrant production, age, and patient outcome in 17 GBM patients undergoing DC immunotherapy [39]. They found that thymic function, as reflected by its ability to produce CD8+ T cells, was directly proportional to good clinical outcomes in mice and human GBM patients and inversely proportional to age. Although patient age has long been a predictor of mortality and prognosis, their findings suggest that it is actually thymic function, which is inversely correlated with age, which may be the more telling factor. Thus, this non-specific immune parameter may later serve as an important prognosticator in glioma immunotherapy.
Caruso et al. conducted a Phase I study of 9 pediatric brain tumor patients undergoing immunotherapy via autologous tumor RNA pulsed DCs [40]. The cohort was comprised of a wide range of different tumor histologies (see Table 1). Although they detected a modest increase in anti-tumor antibodies in some patients, they did not appreciate any increase in T-cell mediated antitumor immunity. This may be explained by their findings that their patients had impaired immunocompetency prior to the start of the trial. Despite this, they reported clinical responses in three patients during the course of their study.
De Vleeschouwer et al. explored the possibility of assessing immunotherapeutic progress through the use of magnetic resonance imaging (MRI) and methionine positron emission tomography (MET-PET) [17]. By monitoring contrast enhancement changes in relation to metabolic uptake ratios they could postulate at which point an immune response had occurred. This group published the findings from their Phase I clinical trial of 12 recurrent malignant glioma patients that were vaccinated with tumor lysate-pulsed dendritic cells [16]. Interestingly, they were the first to induce DC maturation ex vivo for glioma immunotherapy based on recent evidence arguing that the injection of mature DCs may mediate a more potent anti-tumor response [41,42]. The extent of resection was stressed in this study as prolonged disease free survival was only achieved in two patients who underwent gross total resection (GTR) prior to vaccination. Moreover, one patient who received only partial tumor resection suffered Grade IV neurotoxicity (National Cancer Institute common toxicity criteria) secondary to vaccination-induced peri-tumoral edema. As such, they argue that maximal resection may help avoid such dangerous complications during CNS immunotherapy. Akin to our conclusions [33], this study further champions the need for maximal resection to improve the potential efficacy of vaccination strategies for malignant gliomas.
In a Phase I/II study of tumor lysate-loaded DC vaccination for malignant glioma, subcutaneous injections of DCs loaded with tumor lysate were administered biweekly for a total of three injections [43]. Elevated IFN-γ mRNA levels in PBMCs, positive cytotoxicity assays, increased peripheral CD8+ CTLs, and increased infiltration of CD45RO+ memory and CD8+ T cells in progressive tumor corroborated a positive immune response. Additionally, this study reported an increased median survival in patients receiving vaccinations (133 weeks) compared to historical controls (30 weeks), further substantiating the viability of DC immunotherapy for glioma.
After their initial Phase I clinical trial, Kikuchi et al. [44] continued their work with human patients through a Phase I/II series modeled after their animal studies involving DC-glioma fusion cell injection with peri-vaccination IL-12 [27]. Fifteen patients were vaccinated intradermally with FCs on a biweekly basis for a total of three injects per course, with IL-12 administration on days 2 and 5 following each injection. Interleukin-12 was given as it had been shown to enhance the antitumor effects of FCs in mouse models. Similarly, they found that treatment efficacy using FC/IL-12 vaccination in human patients was better than FCs alone. Although, they were able to demonstrate cellular anti-tumor immunity in only a few of their patients, they observed much improved clinical outcomes, including four partial responses and one mixed response as determined by imaging. Patients tolerated the treatment regimen well and there were no reported signs of autoimmunity despite the use of systemic IL-12.
Walker et al. investigated the interaction between chemotherapy and dendritic cell vaccines in their Phase I clinical trial [45]. Thirteen patients with malignant glioma were treated with 6 biweekly injections (and every 6 weeks thereafter) of DCs pulsed with autologous irradiated tumor cells. Immunologically, they were able to appreciate an antitumor response by the presence of increased cytotoxic and memory T cells on post-vaccination resected tumor. Of the 8 patients that received adjuvant chemotherapy in addition to immunotherapy, 5 were reported to show objective radiological response to treatment, including one patient who had a complete response. This mirrors findings by Wheeler and colleagues [46] who through retrospective analysis determined that patients who received chemotherapy following DC immunotherapy did better in terms of overall survival and time to recurrence than patients who received either one alone. Although it was previously believed that chemotherapy and immunotherapy were antagonistic forms of treatment [47], this and other studies have added to the accumulating evidence that these two therapies may in fact be synergistic in nature.
In a recent paper, De Vleeschouwer et al. published an update of their work on DC immunotherapy for brain tumors, including 56 patients with recurrent glioblastoma [14]. Patients received intradermal injections of mature DCs pulsed with autologous tumor lysate according to three vaccination schedules that varied in regards to frequency of injections and the presence or absence of tumor lysate boosts (Table 1). In addition, DTH was assessed in 21 patients from which enough tumor material could be removed for appropriate testing. The treatment regimens were well-tolerated with the exception of one patient who developed vaccination-induced Grade IV neurotoxicity as was mentioned in their previous study [16] and two patients who experienced Grade II transient hematotoxicity. Analysis of patient survival and time to progression revealed that gross total resection prior to vaccination was the only independent predictor of progression free survival. Younger age (<35) and GTR were predictive of better overall survival, however, only in univariable analyses. Although it did not reach statistical significance, the regimen that included frequent vaccinations with tumor lysate boosting seemed to have improved PFS. Interestingly, DTH reactivity was not shown to have any correlation with clinical outcome.
Recently, Wheeler et al. reported on their Phase II trial in which they treated 34 patients with new or recurrent glioblastoma [48]. Patients received a total of 4 subcutaneous injections of autologous tumor lysate-pulsed DCs on weeks 0, 2, 4 and 10. Primary outcomes of interest were time to progression and time to survival (TTS). Immunological responses were quantified through measuring the differential expression of IFN-γ mRNA in lysate pulsed DCs expanded from PBMCs collected before and after vaccination. Using normalized IFN-γ production values as previously reported [49], 17 of the 31 patients tested showed a positive vaccine response (≥1.5 fold expression) after 3 vaccinations (responders). The magnitude of increased IFN-γ expression correlated logarithmically with TTS, however, only in vaccine responders. This finding was striking in that it was the first immunological predictor of immunotherapy outcome to achieve statistical significance, likely due to the large number of vaccine responders in this trial. Clinically, vaccine responders had significantly longer TTS (642 ± 61 days) compared to nonresponders (430 ± 50 days). Moreover, disease free progression in vaccine responders was improved by 4.5 months, with responders and non-responders having TTPs of 308 ± 55 days and 167 ± 22 days, respectively. It should be noted that these trends were not significant in patients with recurrent glioblastoma, only in those with newly diagnosed tumors. Finally, it was found that patients in this study experienced a 186-day to 190-day increase in TTP when the course of DC injections was followed by adjuvant chemotherapy, compared to DC therapy alone. This treatment effect was observed indiscriminately between responders and nonresponders, with differences only appreciable when comparing patients with 5-fold IFN-γ increase and all others. These findings supported recent data suggesting that chemotherapy may possibly potentiate the clinical effects of DC-based immunotherapy [46,47].
Current Status of DC Vaccines for Brain Tumors
Safety
Dendritic cell immunotherapy for brain tumors, throughout the 16 different clinical trials and over 200 patients treated to date, appears to be well tolerated across all variations in treatment protocols. A notable exception was one patient who experienced a Grade IV neurotoxicity following DC administration, which was felt to be due to peri-tumoral edema from the gross residual tumor [14]. Another patient, interestingly, developed a subcutaneous glioblastoma with single lymph node involvement following DTH testing [43]. Despite these outliers, most groups have predominantly reported Grade I and II toxicities in response to DC vaccine administration, with no treatment-associated deaths or permanent neurological defects. The most common reason for discontinuing DC-immunotherapy was tumor recurrence/progression, as would be the case with any other treatment modality for glioblastoma. Overall, the relative lack of serious adverse effects supports the safety of DC-based immunotherapies when used in the management of brain tumor patients.
Measures of outcome
One of the major criticisms of immunotherapy has been the lack of evidence supporting its objective clinical benefit (via MR imaging response criteria) despite the numerous studies that have validated its immunologic anti-tumor response [50]. However, this assertion was posed while evaluating clinical outcomes of immunotherapy using antiquated imaging criteria, which many now argue may not be an appropriate means of assessment in the presence of improved imaging technologies and greater emphasis on disease control/stability, quality of life, and overall survival [45,51,52]. Moreover, although systemic evidence of an anti-tumor responses following dendritic cell immunotherapy has been demonstrated on many occasions both in vitro and in vivo, its correlation with actual tumor lysis in human patients is inconsistent at best.
Several groups have tried to determine an immunologic correlate of clinical efficacy in their Phase I/II studies, including measures such as delayed type hypersensitivity (DTH) [14–16], the presence of tumor infiltrating lymphocytes (TILs) [33,34,43–45], and antitumor immunity in vitro from systemic CTLs [15,32–34,36,37,43]. Results have been mixed, particularly with DTH which was only shown to be predictive of improved survival in one study to date [15]. Tumor infiltrating lymphocytes (TILs) in relapsed tumors and systemic antigen specific CD8+ anti-tumor T cells following vaccination are ubiquitously found in patients that seem to respond to immunotherapy; however, they are not prognostically predictive as many non-responders present with such cells as well. It is thought that the microenvironment of the tumor itself, correlated with immunosuppressive cytokine release (e.g. TGF-β), may inhibit the exacting of actual tumor killing despite sufficient cellular immunity [33,36]. Questions regarding which biologic indices are predictive of clinical outcome will continue to be elucidated as larger cohorts are investigated in multi-center Phase II clinical trials for glioblastomas (e.g., DCVax™). Presently, time-to-progression (TTP) and overall survival (OS) remain the best measures of efficacy in dendritic cell immunotherapy.
Methods of Dendritic Cell Vaccine Development and Administration
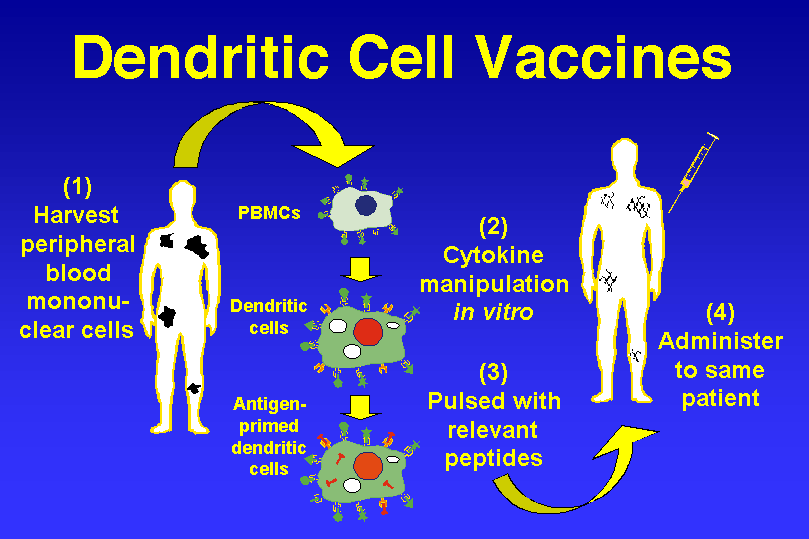
Notwithstanding a decade of use, there still remains a great degree of variability in the development and administration of dendritic cell vaccines. Only a few studies have systematically examined these differences, resulting in a lack of data regarding the most effective means through which to carry out DC-based immunotherapy for CNS neoplasms. Some of these specifics have been resolved on account of information obtained from animal models or through empirical evidence gleaned from common practice. For example, although there are several different methods through which dendritic cells may be acquired, in all of the clinical trials involving DC immunotherapy for glioma to date, they were exclusively manufactured through the differentiation of peripheral blood mononuclear cells (PBMCs) ex vivo. There now exist many methods through which DCs can be produced efficiently and in large enough quantities for clinical trials [53–56].
Similarly, no studies exist comparing the efficacy of different sources of antigens in propagating anti-tumor immunity in human patients. The vast repertoire of antigen-loading strategies includes whole tumor cells [27,35,44], apoptotic tumor cells[45], acid-eluted tumor peptides [19,32–34], synthetic peptides [20,21], tumor lysate [14–17,21–26,38,39,43,48], and tumor cDNA/RNA [28,36,40,57]. The effectiveness of these methods in stimulating DC-mediated antitumor immunity has primarily been studied in animal models for proof-of-principle rather than comparative analyses. Although some animal studies have evaluated the efficacy of different sources of antigens in stimulating a DC-mediated anti-tumor response [21,28], the choice of antigenic stimuli in clinical trials seem largely based on previous work with pre-clinical models and theoretical considerations. Clearly, prior experience with a particular DC vaccination protocol allows for ready transition from bench to bedside. Theoretically, however, methods utilizing a wide range of autologous tumor antigens have been favored over peptide selection. This allows the vaccine to target all tumor associated antigens without requisite characterization, helping avoid clonal selection of antigen-loss variants and subsequent tumor-escape [58]. Choice of antigen must also be considered for pragmatic reasons, as poor availability of resected tumor tissue may favor the use of cDNA/RNA to pulse DCs as these antigens are readily amplified through molecular techniques [9]. Still, as most of the antigen sources available to immunotherapy have been shown to prime dendritic cells appropriately, their use may remain largely an empiric choice until future studies comparatively examining their functionality and practicality are conducted.
It is well accepted that antigen loading is most effective when pulsing phenotypically immature dendritic cells. However, the maturation state in which to administer DCs to patients following this step remains unclear. Numerous studies have shown that dendritic cell maturation is necessary for effective DC migration [59] and T-cell stimulation [42,60], thus making them more effective in generating an anti-tumor response [15]. Given the need for an inflammatory stimulus or cytokine to induce dendritic cell maturation, DCs have been matured ex vivo in order to theoretically ensure proper functioning once they reach the lymph nodes of the host [14–17]. This is supported by work by Yamanaka et al. who found that patients receiving mature DCs experienced a greater overall survival than patients receiving immature ones [15]. However, Barratt-Boyes et al. were able to demonstrate that immature antigen-pulsed dendritic cells undergo natural maturation when injected intradermally and are quite capable of stimulating appropriate anti-tumor T-cell pathways in vivo. Moreover, they argue that the administration of immature DCs may even be superior to that of mature DCs, as the latter have relatively decreased emigration rates from the injection site [18]. As clinical trials have demonstrated clinical benefit with DC regimens using both immature and mature dendritic cells, future studies comparing the two preparations will be needed to further evaluate the effect maturation status has on clinical efficacy and patient survival [61].
The frequency of DC injections in clinical trials was initially modeled after administration schedules found to be effective in immunotherapy for non-CNS tumors [19]. Since then, the majority of studies have roughly followed a biweekly injection regimen, with number of vaccinations varying from 1 to 22 times (Table 1). Given the many differences in other aspects of the vaccination protocol, it is difficult to compare the efficacy of DC administration frequency between published studies. It has been argued that vaccination should be given expediently following maximal surgical cytoreduction, chemotherapy, and/or radiotherapy in order to fully benefit from the rebound in immune function following gross total resection prior to tumor recurrence [52]. Although early initiation of DC immunotherapy is encouraged, data from animal [26] and patient [14] studies suggest early follow-up vaccinations are not as critical, and in fact may hinder the immune response by causing activation-induced death of recently activated T cells. Instead, these studies have demonstrated that booster injections with tumor lysate alone may be more beneficial in stimulating an anti-tumor response. Interestingly, many studies have employed the testing of delayed type hypersensitivity (DTH) using tumor lysate [14–17,37,48], which may have inadvertently served as a form of booster and improved anti-tumor immunity. Given the lack of controlled studies addressing these issues, the timing and frequency of DC vaccine administration remains largely based on empirical experience, and will require future studies to determine an optimal schedule.
The optimal dose of dendritic cells has similarly been questioned. Even from early pre-clinical studies, it was evident that low inoculations of DCs could stimulate an anti-tumor response [16]. Dose escalation protocols in clinical trials have substantiated the finding that DC-mediated immunity is an “on/off” rather than a dose-response phenomenon, as increasing numbers of these APCs do not affect the magnitude of the CTL response [14,33]. This is reassuring, as large quantities of autologous tumor lysate-pulsed dendritic cells were sometimes difficult to obtain during dose escalation protocols [33,40].
Finally, the route of DC administration best for immunotherapy is still under investigation as well. Dendritic cells can be administered through a variety of ways, including subcutaneous, intradermal, intralymphatic, intranodal, and intratumoral injections. Several studies in mice and non-human primates have examined the differences in lymph node accumulation and T-cell stimulation with each route. Radioisotope tracing studies have shown that intravenous DC administration results in the accumulation of dendritic cells within the spleen and liver. However, interestingly this method results in the greatest humoral anti-tumor response as indicated by increased tumor antigen specific antibodies [62–65]. Conversely, intradermal [62,65], intralymphatic [62], intracranial [66], intranodal [59], and subcutaneous [63] injections of DCs have been shown to drain to lymph nodes and induce greater T-cell mediated immunity against tumor antigens compared to intravenous injections in pre-clinical models. Much attention has been given to the intranodal or perinodal administration of DCs, as lymph nodes are acknowledged as the processing centers responsible in mediating antigen presentation and T-cell activation [67]. Some investigators have questioned how the placement of these injections may alter the potency of the immune response. Recently Calzascia et al. were able to show that the distance from the cervical nodes was not as critical as the location of the tumor itself [68]. Although there was some improved tissue tropism for the CNS when DCs were administered into cervical lymph nodes, they found that the ultimate determinant of homing signals was the residence of the actual tumor, as was evidenced by CNS-tropic T cells following inguinal node DC injection in an intracranial tumor model. To date, only one clinical trial has investigated the differences in patient outcome between two injection routes. Yamanaka et al. found that patients who received both intratumoral and intradermal DCs had prolonged survival compared to those that received intradermal DCs alone [15]. Further studies comparing injection sites and modalities in inducing antitumor immunity are still needed.
Patient selection for Dendtic Cell Vaccines
The increasing volume of studies reporting on the clinical response to dendritic cell-based immunotherapy has allowed for the analysis of patient demographics to better determine who may benefit the most from this novel treatment modality. As with traditional therapies for malignant brain tumors, younger patients (<40 years old) receiving DC vaccines appeared to do better in terms of overall survival compared to older patients with similar tumor histologies [48,69]. Though this may in part be attributable to the general trend for younger glioma patients to have better prognoses, one particular study was able to demonstrate that this was primarily due to associated declines in thymus function with increasing age [39]. They maintained that CD8+ T-cell production from the thymus was a prognostic indicator of response to DC immunotherapy independent and superseding that of patient age.
Another critical patient characteristic that seems to have an effect on clinical outcome is surgical management. Those patients who underwent gross total resection of their brain tumor experienced significantly better progression free survival (PFS) compared to otherwise similar patients with appreciable residual tumor [14]. Moreover, bulky residual tumor or active tumor recurrence at the time of vaccination appears to debilitate the anti-tumor CTL response [33].
Finally, patients with newly diagnosed malignant glioma seem to achieve greater response rates than those with recurrent tumors [48]. Although these findings remain to be validated by future studies, it would appear that younger patients with newly diagnosed malignant glioma that are amenable to gross total resection would stand to benefit the most from DC based vaccines.
Synergy of Dendritic Cell Vaccines with other therapies
Although much progress has been made in DC-based immunotherapy for CNS tumors, objective clinical responses for vaccinated brain tumor patients remains inconsistent. Consequently, some groups have examined the use of adjuvant treatments to augment the effects of dendritic cell vaccination. These methods include adjuvant chemotherapy, cytokine administration, and toll-like receptor (TLR) agonists.
The use of cytokines to supplement DC-based immunotherapy in human patients is an extension of work done in pre-clinical animal studies [24,27,29]. Although systemic cytokine administration has only been used in one DC vaccine clinical trial to date [44], studies conducted in vitro as well as with human patients have shown that cytokines such as IL-10 [70], IL-18 [71], and IL-23 [72] may enhance the immune response of effector cells in DC immunotherapy. As the data regarding this adjuvant modality is scarce, further studies are needed before routine clinical use of systemic cytokines can be considered.
The use of standard treatments such as chemotherapy to aid immunotherapy has been considered as well. Chemotherapy has traditionally been regarded as an antagonist to the treatment effects of immunotherapy, because of its effects of bone marrow suppression causing lymphopenia. There has also been a belief that the dead apoptotic tumor cells would produce immune tolerance, exacerbating the lymphopenic state that results as well. However, mounting evidence argues that these apoptotic tumor cells may provide a rich antigen source for dendritic cells and that prompt DC vaccination following chemotherapy may actually provide greater benefit than delaying treatment [47]. Recently, studies have shown that when chemotherapy is used adjuvantly with DC-based immunotherapy, patients experience prolonged overall survival as well as increased time to disease progression [45,46,48]. As evidence suggests that chemotherapy in the setting of DC immunotherapy may actually be beneficial rather than obstructive, it may be prudent to further investigate multi-modality treatment strategies for the simultaneous treatment of brain tumor patients.
Conclusion
Over the past decade, dendritic cell-based immunotherapy for CNS tumors has progressed from pre-clinical rodent models and safety assessments to Phase I/II clinical trials in over 200 patients, which have produced measurable immunological responses and some prolonged survival rates. However, many questions regarding the methods and molecular mechanisms behind this new treatment option remain unanswered. Results from currently ongoing and future studies will help to elucidate which dendritic cell preparations, treatment protocols, and adjuvant therapeutic regimens will optimize the efficacy of DC vaccination. Additionally, it will be important to characterize the pathways underlying the immunosuppressive microenvironment of brain tumors that currently hinder anti-tumor responses. Combined with further advances in the manipulation of various lymphocyte subsets such as regulatory T cells and NK-like T cells, in addition to the usual armament of CD4+ & CD8+ T cells, understanding these immunologic intricacies will help maximize the cellular efficiency of immunotherapeutic techniques. As clinical studies continue to report results on DC-mediated immunotherapy, it will be critical to continue refining treatment methods and developing new ways to augment this promising form of glioma treatment.
Heat Shock Protein (HSP) is a chaperone protein that helps form proteins, stabilize cells against heat stress and aids in the degradation of proteins. HSP is expressed in response to a rise in body temperature or stress levels elicited by such as infection, inflammation or injury. HSP plays a strong role in antigen presentation, which increase the efficacy of vaccines, including anti-cancer vaccines.
Hsp90, a type of HSP, can stabilize proteins required for tumor growth, which is why Hsp90 inhibitors are being investigated as anti-cancer drugs. By inhibiting Hsp90, the tumor cells become unstable and begin to die. Clinical trials of an anti-cancer vaccine are underway and many in the scientific community hope a viable anti-cancer vaccine will become a reality.
Sources and Research:
